Assembling the Tree of Life | By Daniel Oppenheimer | March 3, 2008
In the beginning
Ed Theriot’s contributions to the Assembling the Tree of Life (AToL) project—“the story of life on Earth, in a nutshell”—often begin pretty humbly.
“Sometimes we go down to Waller Creek, right on campus, with a turkey baster and a toothbrush, and we suck up algae from the water and scrub it off the rocks,” says Theriot, the director of the Texas Natural Science Center and a biology professor at The University of Texas at Austin. “We recently found what looks to be a new species of diatom that way.”

From there, the process gets fancier. In the lab, individual diatom cells are plucked one by one from the water sample and placed into tubes of liquid culture media, where they grow by cell division into millions of copies of the original cell. From this batch of cells, the DNA is chemically extracted and individual genes are sequenced. Genetic sequences from similar species are aligned and translated into a form that computers can interpret. These sequences are then run through statistical algorithms that consider millions, billions and even trillions of possible evolutionary (“phylogenetic”) relationships between the species before selecting the one that best explains the similarities among all of the sequences.
The trees are further refined by looking at the morphology of the species—what they look like—along with all the other sources of non-molecular information that illuminate what they eat, how they store energy, how they reproduce and how long they’ve been on the planet. At the end of it all, if a biologist like Theriot is lucky, there’s a useful and pretty accurate representation of when, over the last few hundred million years, hundreds or thousands of species of diatoms diverged from each other and went their separate evolutionary ways.
All this is only a small part of the multi-institutional, interdisciplinary, multi-million dollar effort—funded primarily by the National Science Foundation (NSF)—to assemble the tree of life. The project will, by the time it’s completed, call on the energies, talents and patience of thousands of biologists, mathematicians, geologists, computer scientists and technicians working together across generations, continents, ecosystems, species, academic cultures and computer languages.
Tens of millions of organisms will be collected, sequenced, described and virtually represented. New algorithms will be developed to sift through and extract, in minutes, the kinds of meaningful analyses and comparisons of DNA sequences that would once have taken even the world’s fastest computers months, years or millennia to produce. New software will be coded. New information databases will be painstakingly constructed and integrated with each other.
Many of the applications that will flow from this project, in which scientists at The University of Texas at Austin are playing a central role, will seem like science fiction.

Within a few decades, patients should be able to walk into their doctors’ offices with a common cold and walk out with a treatment that’s been cooked up, on the spot, to combat the particular strain of cold that’s afflicting them. Within a hundred years, biologists may be able to play with the design of life as easily as Apple plays with the design of its laptops.
More simply, the AToL project will realize a founding dream of modern biology. It’s the “tree of life” that Charles Darwin envisioned when he published “The Origin of Species” in 1859—a nearly comprehensive family tree showing how a single organism evolved into the tens of millions of distinct species that now populate the Earth.
“The tree of life is sort of the holy grail for all of biology,” says Theriot, “and it has ramifications for almost everything, from medicine to ecology to agriculture.”
In the field
Along with Theriot, seminal work on assembling the tree of life is being done at the university by biologists Robert Jansen, David Hillis, Randy Linder and David Cannatella, computer scientists Tandy Warnow, Warren Hunt and Daniel Miranker, geologists Tim Rowe and Julian Humphries, and dozens of graduate and postdoctoral students working with these faculty.
It’s a project, says Jansen, that was barely conceivable a generation ago.
“It was at about the time that I got my Ph.D., in 1982, that people were beginning to seriously consider new approaches to phylogenetics,” says Jansen, who was principal investigator on the AToL project that just published a tree relating all the major groups of flowering plants. “Prior to that, we would just draw a tree and say, ‘This is how these species are related,’ and not really provide evidence.”
Jansen, who’s also working with Theriot on the algae project, created his first phylogenetic tree 25 years ago for his doctoral thesis. He assembled that tree, which dealt with a few dozen species of sunflower-like plants common in Mexico, based entirely on a visual examination of the morphology of the different species of plants. He extracted no DNA, did no multiple sequence alignment and used no sophisticated algorithmic software.
For his recent flowering plant project, Jansen and his colleagues sequenced the entire genomes of the chloroplasts (the photosynthetic hearts of plant cells) of 64 species of plants. The genetic information they amassed was so great, running to millions of nucleotides (the molecules that make the code in DNA), that they had to create new software to align it. Since then, says Jansen, even better sequencing techniques have been developed.
“You can now get 100,000,000 nucleotides in four hours,” he says.
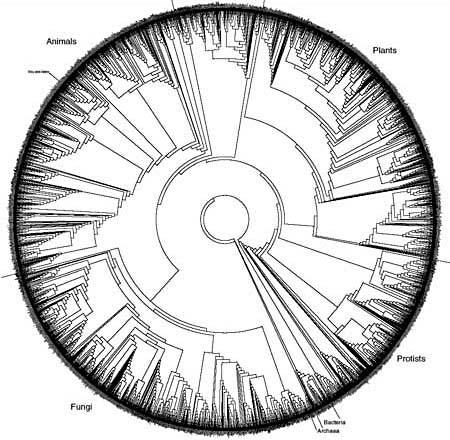
“This wasn’t feasible until the late 1980s and early 1990s, when several things came together,” says biologist Hillis. “We developed the ability to rapidly collect molecular data—to rapidly sequence genes. The computer revolution allowed us to deal with such a massive volume of information. And we developed the analytical methods to put those two things together.”
Hillis and biologist Cannatella are on the steering committee of the “AmphibiaTree” group, which has done groundbreaking work clarifying the phylogeny of modern frogs and salamanders.
They’re doing the kind of heavy biological lifting that will, over the next few generations, make it possible for us to fill out most of the picture of extant life—taking us from the roughly 1.7 million species now identified to the estimated 10-100 million species that co-exist with us on the planet.
“Most of the species on the planet aren’t even known yet,” says Hillis, “and without the tree of life, there’s not a simple way to take information about a new organism that someone discovers and put it in a context of all the rest of life.”
In the late 1990s, Hillis helped convene one of the three workshops that led directly to the NSF’s funding of the AToL project, and since then he’s stayed involved in spreading the word about the tree of life. He oversees a grant to train graduate students in computational phylogeny, and he’s created, with his colleagues Robin Gutell and Derrick Zwickl, what’s perhaps the most pedagogically useful—and almost certainly the coolest-looking—picture of a branch on the tree of life [PDF].
Much of the research Hillis, Jansen, Theriot and their colleagues are doing isn’t so different, at the laboratory level, from what they were doing before the first AToL grants were awarded in 2001. The large-scale organization that the project is facilitating, however, has sped up the process, and brought into focus how valuable the collaboration across disciplines can be.
In the matrix
For Warnow and Linder, a computer scientist and a biologist, respectively, the synthesis of insights from the different disciplines guides them as they deal with what is perhaps the greatest analytical challenge of the AToL project—the near-infinitude of the possible evolutionary trees.

So rather than solve these problems in any final way, Warnow and Linder and other scientists working on this aspect of the problem are forced to generate new and better ways to estimate the best possible evolutionary tree (or trees) for a given group of species.
Refining their estimations entails, among other things, writing statistical algorithms that hop through the total realm of all possible evolutionary trees—“treespace,” as they call it—in a balanced way. The journey must be random enough to benefit from the great diversity of potential trees but still directed enough to recognize that if a given tree seems pretty promising, it’s worth generating similar trees to see if they’re even better.
Biology-based models are also important in estimating a good tree. For example, the tree must make sense with what we know about the fossil record and the geographical dispersal of the different species, and it must fit with what the most sophisticated models of evolution tell us about which kinds of mutations occur more frequently than other kinds of mutations.
At the same time, Warnow and Linder’s software takes into account that even the basic molecular data sets that are being analyzed by the algorithms—the similar strings of nucleotides from similar species—can generate radically different trees depending on how they’re fed into the algorithms in relation to each other.
To deal with this “multiple sequence alignment” problem, Warnow and Linder generate likely histories of how and when comparable genetic sequences (as opposed to species) mutated away from each other. By adding this uncertainty to the basic uncertainty of the tree estimate process, the computational problem becomes even greater. But it’s worth it, says Warnow, because the results are potentially so much more accurate.

In the archives
Warnow and Linder’s generation of potential phylogenetic trees creates the opportunity for computer scientist Hunt’s involvement with AToL.
Hunt and one of his graduate students, Serita Nelesen, have found a vastly more efficient way to store and analyze the terabytes of information the phylogenetic analyses produce. They recognize that in a typical analysis, the many possible evolutionary trees share large pieces of structure, only differing in some of the details.
“Instead of reproducing each part of every tree when you store it, which is what’s been done up to this point,” says Nelesen, “we have a way to replace the redundant parts with a kind of digital pointer that says, ‘This is exactly like that section of tree over there.’ It saves an amazing amount of space.”
That this problem hasn’t been solved before, says Hunt, is a function of the fact that the size of the groups of species being analyzed has, until recently, been pretty small. There just hasn’t been as much data to analyze.
Also, the code has been written mostly by biologists who don’t necessarily have the luxury, or the professional inclination, to peer into the future of data storage and analysis, when the scale will involve assimilating millions of species into a single tree.
“As computer scientists,” says Hunt, “we can do it in a way that’s more industrial grade going in. We can design algorithms with scaling in mind.”
In the mix
For Miranker, who’s creating database software to store two- and three-dimensional images of organisms, the question is less about compressing data than it is about organizing it.
Miranker’s software includes features that are common in commercial applications but rarely available in scientific software. Whenever users sit down to enter the (very long Latin) taxonomic names of their samples, for example, the system suggests autocompletions, thus reminding the scientists of the community’s preferred names and reducing the incidence of typos. The benefit, says Miranker, is greater convenience for the scientists and greater precision for the database.
More significantly, from a phylo-informatic perspective, the software enables better search results. By making the process easier, biologists will upload more images and annotate their images more consistently and thoroughly. The database should then grow richer and more accurate as time goes on, and the various searches for a particular species—or bone, or cellular structure—more productive.
By designing software that imposes certain standards on its users, Miranker has had to wrestle with the future of biology and with how much is at stake in decisions—like what name to assign to a particular species—that may seem uncontroversial to an outsider.

“I’m collecting from the biological community what they think their requirements are, and I’m trying to create a piece of software that helps them do their job right in as painless a way as possible.”
In the mind’s eye
Geologist Rowe’s work with the Digital Morphology library (DigiMorph.org)—a digital archive of 2-D and 3-D images of both living and extinct species—is a reminder that when assembling the tree of life, scientists are still working with whole organic species, rather than just strings of genetic data.
“There’s a sense out there that with all the new molecular methods, morphology is fading, but really it’s undergoing a renaissance,” says Rowe, the project director of DigiMorph and one of the managers of the university’s high-resolution X-ray computed tomographic (X-ray CT) scanner, which has done many of the scans that now reside at DigiMorph.org.
It’s not just that the large skeletal canal revealed by a deep scan of a 120-million-year-old proto-platypus fossil can resolve a phylogenetic controversy. It can, as Rowe discovered recently. His scan of a Teinolophos fossil revealed that the platypus and its closest extant relative, the echidna, diverged tens of millions of years earlier than was previously thought.
It’s also true that the hard morphological facts documented by scans can undermine, as Rowe’s platypus-scan did, entire sets of assumptions about the nature of mammalian evolution.
“It underscores the importance of fossils in testing the assumptions we make when speculating from molecular data,” says Rowe.
The images, and the ways that Rowe and his colleagues are engineering them, can also be powerful tools for organizing information. Images can be annotated. They can be supplemented with other kinds of information about the species. They can link to other Web sites. And they can even, in one of the cooler applications that DigiMorph offers, be manufactured into actual, physical objects made of plastic that can be handled and studied as if they were the original specimens.
In the end

“Individual genes, organisms, populations, species—they’re not interchangeable in the way that in chemistry, for instance, a hydrogen atom is always a hydrogen atom,” he says. “In order to make biology predictive, we have to understand the history and the relationships.”
The goal is also, of course, to complete the tree, or come as close to finishing it as possible. (Because new species are always evolving, scientists can only hope to get close to completing the tree.) And it’s to provide a fundamental knowledge base—a kind of periodic table of the elements for biology—with which to revolutionize applications in medicine, agriculture, ecology and other fields whose connection to biology hasn’t yet become clear.
The purpose is also, perhaps, a little bit spiritual—to commune in the presence of the mystery of life.
“The part of the labwork I love the most,” says Theriot, “is actually looking at the diatoms through a scanning electron microscope. The shells are incredibly beautiful and ornate. Every time I do this, I’m sure I’m seeing something that no one’s ever seen before.”