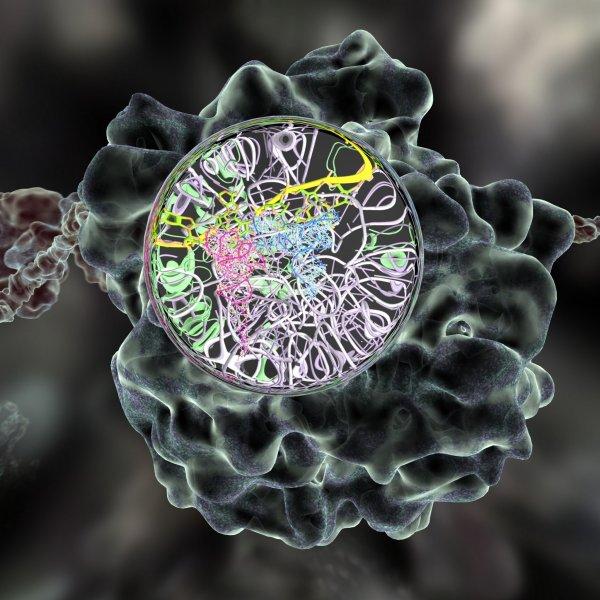
A merged multi-scale structurally valid visualization of the ribosome; the green volume occupying model and the tertiary and secondary structural model is obtained from reconstructed single particle cryo-electron microscopy, while the atomic-resolution structures is from X-ray crystallography resolved models.
Gene-editing or genome engineering is the altering of DNA within a living organism. Once believed to be far-fetched and unthinkable, it is becoming more and more common due to scientific breakthrough techniques like CRISPR. What most people don’t know though is the use of computing tools in conjunction with CRISPR make gene-editing as efficient and mistake-free as possible—making it a viable cure to deadly genetic diseases.
Since its initial discovery in the late 90s, scientists have been studying CRISPR or clustered regularly interspaced short palindromic repeats of DNA inside bacteria. They found that bacteria protect themselves from viruses by using a protein called Cas9 which is guided by the CRISPR sequence to remove corrupted or viral DNA.
Today, scientists have gained enough understanding to develop CRISPR in labs and manipulate it to edit the genes of a variety of organisms. It is a powerful tool that many see as a potential cure to serious genetic diseases such as cancer, HIV, and Huntington’s disease. With the ability to target and edit highly specific segments of DNA, CRISPR is essentially able to tackle sickness right at its roots.
Theoretically, gene-editing should behave like the autocorrect feature of many word processors, replacing problematic genes with healthy ones in the way that typos are corrected. However, this process is highly complex and oftentimes imperfect. CRISPR-Cas9 molecules may sometimes target the wrong genes, replacing healthy segments of DNA with those that do not belong there.
As Texas Computer Science and Integrative Biology Professor William Press says, “the Cas9 molecule finds a place and binds, but this is only useful if it’s the only place to bind”. In other words, the Cas9 molecule can be unpredictable and when the stakes in gene-editing are this high, making a single mistake could lead to undoable, catastrophic, and even life-threatening results.
Computer scientists at UT are working hard to change this. They have developed a way to understand the kind of errors that CRISPR might make. This method, called the Chip Hybridized Affinity Mapping Platform (CHAMP) directly measures which genomes and DNA segments CRISPR will interact with. At its core, it is a genome sequencing chip. According to Press, “before CHAMP, finding binding sites was all guesswork—you would have to try each binding site experimentally in the lab, one at a time, each one oftentimes a full day of work...but with CHAMP—you can easily test a million matches at a time.”
The whole project is open source, from the designs for a 3-D printed mount that holds the chip under a microscope to the software that the team developed for analyzing collected data. As a result, other scientists and researchers can easily replicate and further build upon this revolutionary technique.
Press says CHAMP provides an experimental and computational pipeline that will enable the important next step: to implement a machine learning approach in which examples of true bindings and off-target bindings are provided to a computer to generalize underlying patterns. Future systems will then be predictive, requiring the CHAMP pipeline only for experimental confirmation.
Even so, identifying bacterial CRISPR sequences with their associated Cas9 like nucleases is one peice of the broader gene-editing puzzle. It is now also possible to design and synthesize short RNA molecules to match specific DNA sequences. These molecules can then guide nucleases and enzymes such as CAS9 to an intended DNA target similar to CRISPR-Cas9 machinery.
“You need to design and create the right RNA for the identified gene sequence you need to edit, and also identify the RNA binding proteins and nucleases that perform the editing” says, Texas Computer Science Professor and Director of the Center for Computational Visualization Chandrajit Bajaj. Bajaj works on the molecular modeling and recognition aspects of the problem, creating tools for determining structure and function models from molecular imaging, molecular docking, and drug discovery.
It turns out, in this area, computers can be used again to simulate and test a multitude of designer RNA models and binding proteins for specific DNA binding sites. “Just as in drug discovery, the search is for the best guide RNA and nucleases that bind and edit DNA sequences with specificity”, says Bajaj. Further, “Can these be synthesized? What are associated nuclease enzymes that can facilitate this DNA binding and gene-editing? Can these also be safely delivered into the body?”
These are some of the many questions that must be asked and answered to realize the full potential of using CRISPR and other CRISPR inspired technologies to combat human diseases. Utilizing computing to simulate binding affinity and optimal molecular recognition are design optimization and machine learning problems just as much as they are biological ones.
To take the guesswork out of CRISPR or designing CRISPR-Cas9 like molecular machinery for gene-editing is an extremely powerful thing—especially when the whole process is ultimately used to remedy currently incurable illnesses. Humans just simply cannot work through all the possibilities and today’s data-driven techniques using machine learning, computational modeling, and simulation are the key enablers to making gene-editing as safe and mistake-free as possible. These tools will continue to empower computer and biological scientists alike to revolutionize the future treatment of genetic diseases.